Exercise solution for Chapter 2, Part 1
As always, load libraries first.
library(ggplot2)
library(tidyverse)
library(dplyr)Exercise 2.3 from Modern Statistics for Modern Biologists
A sequence of three nucleotides codes for one amino acid. There are 4 nucleotides, thus \(4^3\) would allow for 64 different amino acids, however there are only 20 amino acids requiring only 20 combinations + 1 for an “end” signal. (The “start” signal is the codon, ATG, which also codes for the amino acid methionine, so the start signal does not have a separate codon.) The code is redundant. But is the redundancy even among codons that code for the same amino acid? In other words, if alanine is coded by 4 different codons, do these codons code for alanine equally (each 25%), or do some codons appear more often than others? Here we use the tuberculosis genome to explore codon bias.
a) Explore the data, mtb
Use table to tabulate the AmAcid and Codon variables.
Each amino acid is encoded by 1–6 tri-nucleotide combinations.
mtb = read.table("example_datasets/M_tuberculosis.txt", header = TRUE)
codon_no <- rowSums(table(mtb))
codon_no## Ala Arg Asn Asp Cys End Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr
## 4 6 2 2 2 3 2 2 4 2 3 6 2 1 2 4 6 4 1 2
## Val
## 4The PerThousands of each codon can be visualized, where each plot represents an amino acid and each bar represents a different codon that codes for that amino acid. But what does the PerThousands variable mean?
ggplot(mtb, aes(x=Codon, y=PerThous)) +
geom_col()+
facet_wrap(~AmAcid, scales="free") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))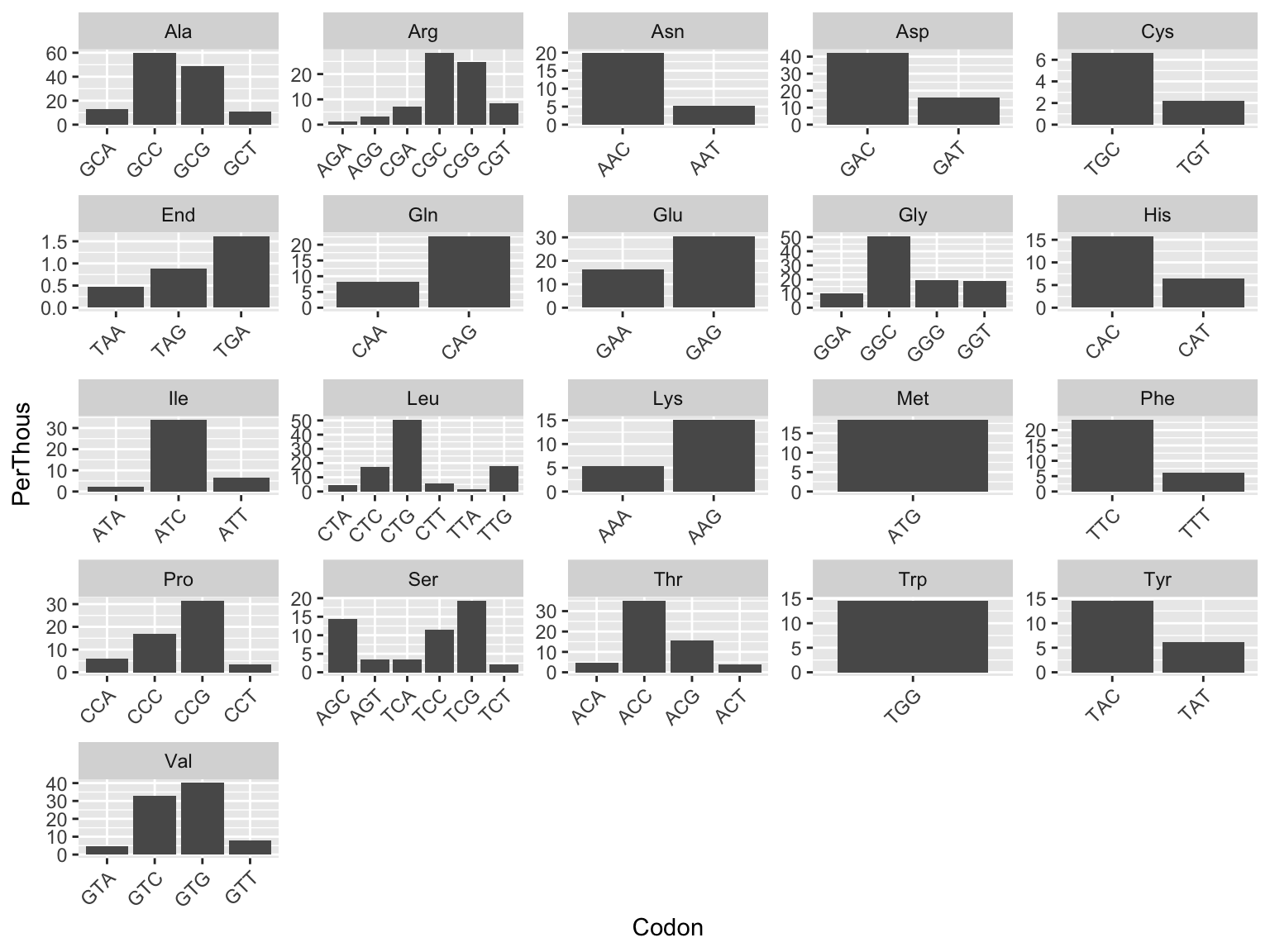
b) The PerThous variable
How was the PerThous variable created?
The sum of all of the numbers of codons gives you the total number of codons in the M. tuberculosis genome: all_codons. Remember that this is not the size of the M. tuberculosis genome, but the number of codons in all M. tuberculosis genes. To get the size of the genome, multiply each codon by 3 (for each nucleotide) and add all non-coding nucleotides (which we do not know from this data set).
all_codons = sum(mtb$Number)
all_codons## [1] 1344223The PerThousands variable is derived by dividing the number of occurrences of the codon of interest by the total number of codons. Because this number is small and hard to interpret, multiplying it by 1000 gives a value that is easy to make sense of. Here is an example for proline. The four values returned align to the four codons that each code for proline.
pro = mtb[mtb$AmAcid == "Pro", "Number"]
pro / all_codons * 1000## [1] 31.560240 6.121752 3.405685 17.032144c) Codon bias
Write an R function that you can apply to the table to find which of the amino acids shows the strongest codon bias, i.e., the strongest departure from uniform distribution among its possible spellings.
First, let’s look at the expected frequencies of each codon.
codon_expected <- data.frame(codon_no) %>%
rownames_to_column(var = "AmAcid") %>%
mutate(prob_codon = 1/codon_no)
codon_expected## AmAcid codon_no prob_codon
## 1 Ala 4 0.2500000
## 2 Arg 6 0.1666667
## 3 Asn 2 0.5000000
## 4 Asp 2 0.5000000
## 5 Cys 2 0.5000000
## 6 End 3 0.3333333
## 7 Gln 2 0.5000000
## 8 Glu 2 0.5000000
## 9 Gly 4 0.2500000
## 10 His 2 0.5000000
## 11 Ile 3 0.3333333
## 12 Leu 6 0.1666667
## 13 Lys 2 0.5000000
## 14 Met 1 1.0000000
## 15 Phe 2 0.5000000
## 16 Pro 4 0.2500000
## 17 Ser 6 0.1666667
## 18 Thr 4 0.2500000
## 19 Trp 1 1.0000000
## 20 Tyr 2 0.5000000
## 21 Val 4 0.2500000Next, calculate the observed frequencies for each codon seen in the data set and use the chi-squared test statistic to determine if the difference between expected and observed codon frequencies is even or if some codon sequences are used more than others.
To start, you can group the data by amino acid and then determine a few things about
the amino acid or the possible codons for it, including the total observations
across all codons for the amino acid (total), the number of codons for that
amino acid (n_codons), and the expected count for each codon for that amino acid
(the total number of observations for that amino acid divided by the number of
codons, giving an expected number that’s the same for all codons of an amino
acid; expected).
codon_compared <- mtb %>%
group_by(AmAcid) %>%
mutate(total = sum(Number),
n_codons = n(),
expected = total / n_codons)
codon_compared## # A tibble: 64 x 7
## # Groups: AmAcid [21]
## AmAcid Codon Number PerThous total n_codons expected
## <fct> <fct> <int> <dbl> <int> <int> <dbl>
## 1 Gly GGG 25874 19.2 132810 4 33202.
## 2 Gly GGA 13306 9.9 132810 4 33202.
## 3 Gly GGT 25320 18.8 132810 4 33202.
## 4 Gly GGC 68310 50.8 132810 4 33202.
## 5 Glu GAG 41103 30.6 62870 2 31435
## 6 Glu GAA 21767 16.2 62870 2 31435
## 7 Asp GAT 21165 15.8 77852 2 38926
## 8 Asp GAC 56687 42.2 77852 2 38926
## 9 Val GTG 53942 40.1 114991 4 28748.
## 10 Val GTA 6372 4.74 114991 4 28748.
## # … with 54 more rowsThe mutate function is used after group_by to do all this
within each amino acid group of codons, but without collapsing to one row per
amino acid, as a summarize call would.
To convince yourself that this has worked out correctly, you can repeat the plot we made before and see that the bars for the expected values are always equal across all codons for an amino acid:
ggplot(codon_compared, aes(x=Codon, y=expected)) +
geom_col()+
facet_wrap(~AmAcid, scales="free") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
Finally, we can calculate the chi-squared (\(\chi^2\)) statistic and compare it to the chi-squared distribution to get the p-value when testing against the null hypothesis that the amino acid observations are uniformly distributed across codons. The \(\chi^2\) is calculated as:
\[ \chi^2 = \sum_i{\frac{(O_i-E_i)^2}{E_i}} \]
where:
- \(O_i\) is the observed value of data point \(i\) (
Numberin our data); and - \(E_i\) is the expected value of data point \(i\) (
expectedin our data)
In our data, we can calculate the contribution to the total \(\chi^2\) statistic
from each data point (in this case, each codon within an amino acid) using
mutate, and then
add these values up using group_by to group by amino acid followed by
summarize to sum up across all the data points for an amino acid.
The other information we need to get is the number of codons for the
amino acid, because we’ll need this to determine the degrees of freedom
for the chi-squared distribution. Next, we used mutate with
pchisq to determine the p-values within each amino acid group for the
test against the null that the codons are uniformly distributed for that
amino acid (i.e., that there isn’t codon bias). These p-values turn out to
be super small, so we’re using a technique to get the log-transform versions of
them instead, which we explain a bit more later. Finally, we used arrange to
list the amino acids by evidence against uniform distribution of the codons,
from most evidence against (smallest p-value so most negative log(p-value))
to least evidence against (although still plenty of evidence against) and added
an index with the ranking for each codon by adding a column with the sequence
of numbers from 1 to the number of rows in the data (n()).
codon_compared %>%
filter(n_codons > 1) %>%
group_by(AmAcid) %>%
mutate(chi_squared = ((Number - expected)^2/expected)) %>%
summarise(chi_squared = sum(chi_squared),
n = n()) %>%
mutate(p_value = pchisq(chi_squared, df = n-1, log = TRUE, lower.tail = FALSE)) %>%
arrange(p_value) %>%
mutate(rank = 1:n())## # A tibble: 19 x 5
## AmAcid chi_squared n p_value rank
## <fct> <dbl> <int> <dbl> <int>
## 1 Leu 135432. 6 -67700. 1
## 2 Ala 75620. 4 -37805. 2
## 3 Arg 72183. 6 -36076. 3
## 4 Thr 58767. 4 -29378. 4
## 5 Val 58737. 4 -29363. 5
## 6 Ile 56070. 3 -28035. 6
## 7 Gly 52534. 4 -26262. 7
## 8 Pro 45400. 4 -22695. 8
## 9 Ser 36742. 6 -18357. 9
## 10 Asp 16208. 2 -8109. 10
## 11 Phe 13444. 2 -6727. 11
## 12 Asn 11404. 2 -5707. 12
## 13 Gln 9376. 2 -4693. 13
## 14 Lys 6382. 2 -3195. 14
## 15 Glu 5947. 2 -2978. 15
## 16 His 5346. 2 -2678. 16
## 17 Tyr 4738. 2 -2373. 17
## 18 Cys 2958. 2 -1483. 18
## 19 End 928. 3 -464. 19As you may notice, these log transforms of the p-values (which we got rather than untransformed p-values in the pchisq call because we used the option log = TRUE) are large in magnitude and negative (so very tiny once you take the exponent if you re-transformed them to p-values) values. If you tried to calculate the untransformed p-values (and we did!), this number is so small (0.00000000e+00) that it is too small for R—it shows up as exactly zero in R, even though it actually is a very tiny, but still non-zero, number. To get around this issue, we told pchisq to work on these p-values as log transforms, and then we left the p-value as that log-transformed value. A group of numbers that are log transformed will be in the same order as their untransformed versions, so we don’t need to convert back to figure out which amino acid had that smallest p-value. We can just sort the amino acids from most negative to less negative using these log-transformed versions of the p-values. We now have the amino acids ranked from most biased codons (1) to least (19).
Sere Williams
Graduate Student in Cellular and Molecular Biology
Seré is interested in plant stress at the genetic level.